近日,吉林大学化学学院、理论化学研究所曲泽星教授/周中军副教授课题组和山东大学刘文剑教授合作在《JACS Au》上发表了题为“Machine Learned Fock Matrix”的研究工作。本研究介绍了一种新型机器学习积分自洽场方法—miSCF,该方法旨在高效预测体系电子结构。
纵所周知,自洽场迭代(SCF)是电子结构计算中最为关键、也是最为耗时的一步。该过程涉及众多积分,如重叠积分、单电子积分和双电子积分,其中双电子积分的计算量为N4(N为基函数数目)。虽然通过双电子积分本身具有八重对称性,可以减少一定量的积分数,但随着基函数的增加,双电子积分的计算量仍然呈现指数增长,为求解Roothaan方程带来了巨大困难。为了解决这一难题,我们提出了一种机器学习积分自洽场方法—miSCF并通过它来构建Fock矩阵。根据
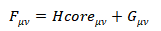
矩阵表达式可知,Fock矩阵与基函数、构型和密度矩阵直接相关。因此,我们构建三种描述符,如积分相似描述符、构型描述符以及角动量描述符,来预测Fock矩阵。为验证miSCF的可靠性,我们测试了若干具有代表性的小分子如氢链和水链以及凝聚态冰结构。结果表明miSCF在能量、波函数和电子密度预测方面均表现出良好性能和可迁移性。这使得仅需少量训练数据即可精确预测体系的各种电子性质,在保持高精度的同时显著降低了计算成本。因此,miSCF方法为量子化学计算提供了高效工具,并为势能面构建、从头算分子动力学和化学反应模拟等进一步应用奠定了坚实基础。
该成果以“Machine Learned Fock Matrix”为题发表在JACS Au(DOI:10.1021/jacsau.5c01200)。吉林大学化学学院、理论化学研究所博士研究生刘鸿材为第一作者,吉林大学曲泽星教授、周中军副教授和山东大学刘文剑教授为共同通讯作者。

论文链接:
https://pubs.acs.org/doi/10.1021/jacsau.5c01200



 当前位置:
当前位置:
